







| Protein: | CG4467-PA |
| Organism: | Drosophila melanogaster |
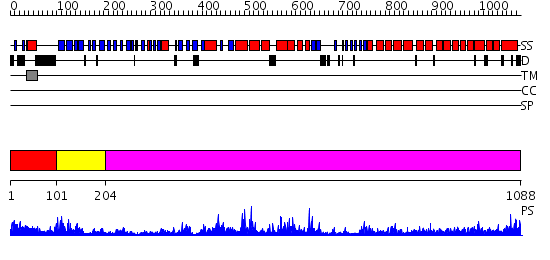
| Length: | 1088 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for CG4467-PA.
| Description | E-value | Query Range |
Subject Range |
|
|
697.0 | [0..1] | [1088..1] |
|
Region A: Residues: [1-100] |
1 11 21 31 41 51
| | | | | |
1 MTNDPDLDDC AFLSGGESSG GRQTRTGVAV CSQRRALLVA GIVLGSLLLT AIIIAYAGPQ 60
61 NDCSCGSKTV SGYETDEENN TQPFNPIATN GEPFPWLEKM
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [101-203] |
1 11 21 31 41 51
| | | | | |
1 LPTSVRPLRY MVTIHPNLTT LDVKGQVTID LHVEKETNFI VLHIQDLNVT EKAIVTPGPK 60
61 GYALKIVKVL EFPPRQQLYI EVKERLKKKS NYTLNLRWYS KLN
|
| Detection Method: | |
| Confidence: | 2.39 |
| Match: | 1hs6A |
| Description: | Leukotriene A4 hydrolase C-terminal domain; Leukotriene A4 hydrolase N-terminal domain; Leukotriene A4 hydrolase catalytic domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [204-1088] |
1 11 21 31 41 51
| | | | | |
1 PEPEGFYVDQ YESSNGVERL LAATVFRPNG ARRAFPCFDE PHVRAPFRIS VFRDRFHIGL 60
61 SNSIVHTTED VGFYMGTGLL RDDFIETPPL PADAVAWVVS DFQRESLQPS AAYIPTTPAP 120
121 PGSGVGGKKS AQLNNYTQLK GKNPPVRNIT ALTHSLNVNL TPRQNLTVVQ PTTTTAWPVN 180
181 LNGNGKPSNL TSLSQSTGSS IKRAPSYTFY APRDLLIRSS FILHTSRDVL EYLQTWLDIS 240
241 YPLTKVDFVA LPSLDRNMIS SLGLVTLKTS FLTDPSSITS EQYQFSALRI AEAMVRQFFG 300
301 GITSRKVLKD VWLWEGLIQY LGIHALAPLQ ETWPLREMYL LKMATAALDI DAIQGWDSIM 360
361 NGTSHDGNNE EFFVQKTAAI FSMLHTAIGE DRFRGCLGSF LKVNRFRTAE PTDLWTICTK 420
421 KANGSKNIKD MMTLWTHQPG FPLLTVTKMG NSISISQRPF RPAEFLAIHD DSYDGNNYNK 480
481 TTLNATDMPS TVATTTQGSK HKVAPHMKWI FPVTYVTDIN NVSETLWMQN VDVTFNVPEN 540
541 VKWIKVNAIQ NGYYRVVYND DNWASLIEEL AANPNRFTSE DRLGMLSDAF TLCHANLLPC 600
601 EITMNMIQYL PSETHYGPMA LALRHLEKWR RILKYSECFL MLSEFIKMKI STVMEKVGWS 660
661 DDGDVATRLL RPEVLLASVL WEDIDSITKA KNMLNQYLYY NGTAIPPNLR EVVYTGSILS 720
721 GEYIYWQHCW ERFVNLQRTS ETFVERMQLL RALGRTKDAW LQNRLLSHVT MLPTEEVVQV 780
781 LKAIAGTPTG GAMACRFLQA KWFELEKRLG PGTISFAKVI SAITQYGATK FDYDELKSLV 840
841 HRFGRGHGMS VLNMTLSSVA SNVEWVARSQ TSLYKWVEGN LHSHR
|
| Detection Method: | |
| Confidence: | 155.0 |
| Match: | 2hpoA |
| Description: | No description for 2hpoA was found. |