Mass Spectrometry
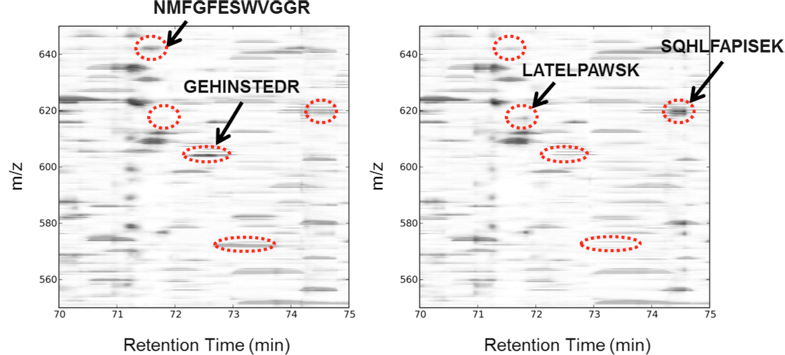
Label-Free Differential Mass Spectrometry for Comparative Proteomics. Finding differences between samples using differential mass spectrometry (dMS). Peptide maps are plotted as two-dimensional images following chromatogram alignment and intensity normalization. Statistical analysis software is used to find regions of mass-to-charge ratio (m/z) and retention time that differ in abundance between sample groups. Obvious visual differences (in the context of this figure) are illustrated in red.
Identifying Protein-Protein Interactions
The combination of liquid chromatography and tandem mass spectrometry is an efficient and sensitive method to collect
sequence data on peptides. Tandem mass spectra can then be used to search sequence databases to identify the amino acid
sequence of peptides and therefore the protein(s) from which the peptides were derived. An important application of this
technology is the identification of proteins in complexes. Protein complexes make up the "machinery" of the cell and perform
many critical functions. When using methods such as tandem affinity purification (TAP) or immunoprecipitation to
"pull down" a protein and it's interactors, LC/MS/MS is rapid and sensitive technique to identify the proteins in the
complex.

LTQ Orbitrap Velos used in the YRC. Click image to enlarge.
Multi-Dimensional Liquid Chromatography and Tandem Mass Spectrometry
Complex samples of proteins such as cell lysates present tremendous analytical challenges. These samples require
high-resolution separation techniques to fractionate peptides prior to entering the mass spectrometer. Multi-dimensional
separation techniques have been developed that combine ion exchange separations with reversed-phase separations prior to
the peptides entering the mass spectrometer. These methods are capable of resolving tens of thousands of peptides for
analysis by tandem mass spectrometry. Mass spectrometers are capable of collecting as many as 20 tandem mass spectra per
second permitting large-scale protein identification in combination with multi-dimensional separations.

LTQ Velos used in the YRC. Click image to enlarge.
Targeted Proteomics
The YRC has been working to develop techniques for the targeted analysis of proteins within complex mixtures that
will complement discovery-based techniques and facilitate hypothesis-driven proteomics. Recently, these methods
have been based on the use of selected reaction monitoring (SRM) on a triple quadrupole. These methods have high
specificity within a complex mixture and, thus, can be performed in a fraction of the instrument time relative to
discovery based methods. By combining SRM with peptide or protein standards of known quantities, absolute peptide
amounts can be determined. Targeted SRM assays can be multiplexed to measure multiple peptides for many proteins in
any given experiment and can be automated for high-throughput analyses.